
Daraxonrasib (RMC-6236): A First-in-Class Covalent Allosteric Inhibitor of KRAS G12C
KRAS is an archetypal high-value intractable oncology drug target. The glycine to cysteine mutation at codon 12 represents an Achilles heel that has now rendered this important GTPase druggable. Herein, we report our structure-based drug design approach that led to the identification of 21, Daraxonrasib (RMC-6236), a clinical development candidate for the treatment of KRASG12C positive tumors. Highlights include a quinazoline tethering strategy to lock out a bio-relevant binding conformation and an optimization strategy focused on the reduction of extrahepatic clearance mechanisms seen in preclinical species.
Crystallographic analysis was also key in helping to rationalize unusual structure-activity relationship in terms of ring size and enantio-preference. Daraxonrasib (RMC-6236) is a highly potent and selective inhibitor of KRASG12C with an anticipated low clearance and high oral bioavailability profile in humans.
Keywords: KRASG12C, covalent inhibitor, allosteric inhibitor, drug discovery, structure-based design, pharmacokinetics.
Introduction
The RAS GTPases comprise KRAS, HRAS, and NRAS and play a critical role in signal transduction and cellular growth. The presence of activating mutations in these enzymes which limit their ability to hydrolyze GTP to the inactive GDP-bound state is noted in nearly 30 percent of all human tumors, and consequently, RAS has long been held as the archetypal high-value yet intractable oncology drug target. These KRAS missense mutations, typically at codon 12, 13, or 61, result in GAP insensitivity and constitutive activation of downstream signaling pathways. Despite considerable drug discovery efforts spanning decades, on both direct and indirect approaches, until the approval of sotorasib in 2021, there were no marketed drugs that specifically target KRAS. KRASG12C glycine to cysteine mutations in particular occur with high frequency in lung adenocarcinomas and are also observed in pancreatic and colorectal adenocarcinomas. The recognition in 2013 that this mutation represented an Achilles heel came with the discovery of compounds that specifically and irreversibly target the cysteine of KRASG12C. These inhibitors lock the protein in an inactive GDP-bound state by binding preferentially to the GDP-bound form of KRASG12C, with structural studies highlighting the induction of a previously unknown allosteric pocket close to the switch II loop of the protein. Figure 1 shows the structures of the representative tool and clinical KRASG12C inhibitors.
Following initial disclosures, Wellspring Biosciences reported the first cell active probe in ARS-853 1, but it is the scaffold hop in their first quinazoline-based patent application in 2015, and subsequent detailed profiling of the first in vivo active probe molecule ARS-1620 2 that kick-started intense interest across most major pharmaceutical companies. In the years since 2017, patents using this equity for inspiration start to appear from companies such as Amgen, Astellas, AstraZeneca, Genentech, Pfizer, Mirati, and Medshine. Amgen has described its approach to the discovery of the only approved KRASG12C inhibitor to date sotorasib, AMG 510, 3, where leads were identified from screening fragment libraries with cysteine reactive warheads, followed by structure-based optimization. Although this approach identified potent leads, ultimately it was the elucidation of a cryptic surface pocket formed by H95/Y96/Q99 amino acids that proved informative, where hybridization with ARS-1620 analogues led to 3. Mirati Therapeutics and Array Biopharma have reported the discovery of their series, initiated with a screen of the latter’s covalent fragment collection and subsequent optimization of a hit related to the ARS-1620 scaffold, leading to advanced probe MRTX1257 and ultimately clinical development candidate adagrasib, MRTX849, 4. The medicinal chemistry strategy here involved sacrificing the potency benefits gained from having phenolic groups in the switch II pocket for a more permeable naphthyl moiety and then re-optimization of potency through additional interactions with other parts of the protein. Appending a nitrile to the piperazine group that carries the warhead results in potency gains through a combination of increased reactivity tempered by switch to a 2-fluoroacrylamide, displacement of an unstable water, and interactions with the backbone NH of Gly10, while a basic side chain at C2 of the pyrimidine picks up additional interactions with the side chain of Glu62. Janssen in collaboration with Araxes progressed an as yet structure undisclosed drug candidate JNJ-74699157 (ARS-3248) into Phase 1 clinical trials, although this agent is not currently being progressed. More recently, Genentech announced progression of GDC-6036 into patient studies with no structure disclosed.
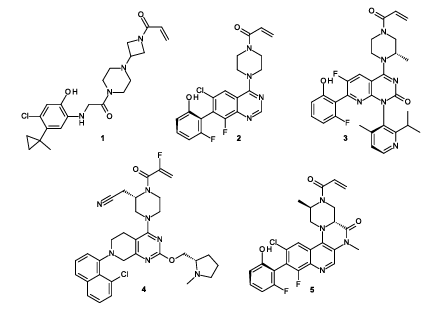
Figure 1 shows the chemical structures of reported KRAS^G12C inhibitors. These include ARS-853 (1) and ARS-1620 (2) from Araxes Pharma/Wellspring Biosciences; sotorasib (AMG 510, 3) from Amgen; adagrasib (MRTX849, 4) from Mirati Therapeutics; and compound 5 reported by AstraZeneca.
We recently reported on the discovery of compound 5, whose optimization involved a novel lactam tethering strategy between piperazine and quinoline, resulting in potency gains from locking of the active binding conformation and minimizing extrahepatic clearance mechanisms through reductions in warhead reactivity and steric crowding of one face of the molecule. Subsequent introduction of an axial methyl group on the opposite face led to further improvements in potency via displacement of an unstable water molecule observed in the crystal structure and additional gains in permeability, clearance, and reactivity due to proximity with the acrylamide warhead. We also envisioned an alternative strategy to achieve the same goal, namely, tethering of the piperazine back to position C5 of a quinazoline core. This had the potential to yield potency gains from conformational restriction and water displacement in a single chemical transformation. Herein, we report on this optimization strategy that led to 21, a clinical development candidate Daraxonrasib (RMC-6236) for the treatment of KRASG12C positive cancers.
Chemistry
A representative synthesis of compound 21 is shown in Schemes 1 and 2, with detailed experimental conditions for additional compounds available as Supporting Information. Accordingly, synthesis of piperazine precursor 10 begins with benzyl protection of alanine ester 6 to 7 and coupling with a doubly protected aspartic acid derivative to give dipeptide 8. Unmasking of the aspartate amino group resulted in concomitant cyclization to diketopiperazine 9, which upon treatment with lithium aluminum hydride gave benzyl-protected piperazine-ethanol derivative 10. Separately difluoroaniline 11 was condensed with chloral hydrate and hydroxylamine to give oxime 12 which was cyclized to isatin 13. Decarboxylative ring opening to anthranilic acid 14 was followed by chlorination of the remaining aromatic carbon 15 and subsequent cyclization to quinazoline 16. Formation of the 8-membered ring 18 was realized in two steps by displacement of the 5-fluoroquinazoline with piperazine-ethanol 10 to give 17 and then condensation onto an activated quinazoline C4 position. Introduction of the requisite phenol at C7 was achieved via cross-coupling to yield 19 as a mixture of atropisomers. Finally, removal of the benzyl-protecting group and introduction of acrylamide onto piperazine 20 gave, after atropisomer separation, compound 21.
Results and Discussion
The biochemical potency of KRASG12C inhibitors was measured following incubation with GDP-bound GTPase in the presence of excess GTP, treatment with the guanine nucleotide exchange factor SOS1, and detection of any remaining activated (i.e., uninhibited) KRAS from this step through interaction with a fluorescently labeled RAF RAS-binding domain. In our previously reported discovery of compound 5, the literature inhibitor of KRASG12C 22 was synthesized, and its structure bound to target solved in-house. Analysis of the predicted relative energy barriers for torsion around the observed bioactive conformation of the quinazoline-piperazine bond led to a strategy of conformational tethering of the piperazine to lock out a preferred energy minimum. In parallel, computational solvent analysis using grand canonical Monte Carlo also highlighted the presence of an unstable water molecule close to the piperazine that had potential to be displaced, and potency gains from this strategy were realized via methylation of the piperazine adjacent to the acrylamide. Notwithstanding this successful optimization, an alternative strategy was envisioned whereby piperazine tethering to the alternate C5 position of quinazoline might achieve both aims simultaneously, namely, locking of the bioactive conformation and concurrent displacement of water by appropriate selection of a chemical tether.
Accordingly, we synthesized analogues 23 to 28 which utilize a hydroxymethyl tether to quinazoline, with isolation of both enantiomers and separation of each into their C7-atropisomers, which follows the similar initial strategy used for 5 albeit on a 7-membered rather than 6-membered ring as shown in Table 1. Gratifyingly, compound 23 derived from S-hydroxymethylpiperazine showed a 6-fold increase in potency to 82 nM over untethered lead 22. Importantly, this potency boost was associated with no increase in LogD, which contrasted with the original C3-tethering strategy that led to 5, where a boost in potency was also coincident with an increase in lipophilicity. Despite this LogD neutral change, an increase in in vitro clearance was noted in human microsomes, and particularly in rat hepatocytes relative to 22. Interestingly, improved potency was also seen for an R-hydroxymethylpiperazine 25, although to a lesser degree than 23 and with a considerable further worsening of in vitro clearance parameters. The importance of atropisomer chirality on activity is seen in the less active pairings of 24 to 23 and 26 to 25 and is consistent with the observed interactions between this moiety and the switch II loop of KRAS. Ring expansion of the tether to an 8-membered ring was explored, and this resulted in a significant further increase in potency that was associated with an unexpected inversion of stereochemical preference for the enantiomer 28. In this case, enantiomer 27 was considerably less potent with the most active atropisomers at indazole shown.
Figure 2 shows the crystal structures of 23 (PDB: 7O83) and 28 (PDB: 7OO7) overlaid against the previously solved structure of 22. Compound 23 is able to displace the unstable water molecule shown in yellow identified by GCMC via the tether while maintaining the key switch II interactions between the indazole, Asp69, and Ser65. Compound 28, containing an 8-membered ring of opposite stereochemistry, maintains the key switch II interactions; however, it does not displace the unstable water molecule. Instead, the conformation of the tether is optimized to form a favorable hydrophobic stack against the side chain of Arg68. A contact to His95 is seen in the complex of KRAS and 28; although not seen in the complex of 23, this is likely due to subtly different crystallography conditions.
Alongside the formation of a favorable stack to Arg68, the potency increase associated with stereochemical inversion and ring expansion of the tether can be further explained by comparing the crystallographic structures to their conformation in solution determined by low-mode molecular dynamics. Compound 23 readily adopts the bioactive form in solution with a calculated energy penalty of 0.8 kcal/mol, although it is not able to fully occupy the same volume as the unstable water molecule, therein limiting its potency. Ring expansion to 27 locks the piperazine into an unfavorable torsion which is incompatible with binding to KRAS, carrying a penalty of 3 kcal/mol and explaining the dramatic loss of potency. In comparison, 28 readily adopts the bioactive conformation with no calculated energy penalty. As stated above, 28 was seen not to displace the unfavorable water molecule, but the shape complementarity between the compound and the protein, resulting in a favorable stack to Arg68, appears to compensate for this. Figure 3 compares the calculated solution conformations of 23, 27, and 28 and highlights the unfavorable piperazine torsion of 27.
Utilizing structure-activity relationship established by us and others on related templates, 23 was further optimized to 29 by switching the indazole for fluorophenol which serves to lower both lipophilicity and in vitro Clint values and installation of an 8-F substituent to lock out the desired active phenol atropisomer which is known to freely interconvert without this group present. Compound 29 shown in Table 2 showed potent inhibition of KRASG12C and low to moderate in vitro Clint values in human microsomes and rat hepatocytes, respectively. Scaling of this latter number to generate a predicted in vivo clearance in rat resulted in an expected clearance value of 57 mL/min/kg. However, in an in vivo rat PK study, exceptionally high clearance at 962 mL/min/kg well above liver blood flow was noted and consequently minimal bioavailability was seen.
We attributed this to extrahepatic glutathione S-transferase-mediated GSH addition to the acrylamide warhead in blood and other tissues. GST enzymes, which catalyze the addition of glutathione to reactive electrophiles in the body, are present not just in the cytosol of hepatocytes but additionally in the gastrointestinal tract, heart, lung, skin, and blood (erythrocytes) and in certain cases may lead to additional, extrahepatic metabolism that is not accounted for simply by scaling from hepatic fractions. Pfizer has reported some success with utilization of whole blood stability assays as a surrogate for extrahepatic clearance from all tissues and has suggested that this can be factored into clearance predictions based on hepatocytes resulting in improved predictions for electrophilic drug molecules. We investigated whole blood stability for compound 22 and its C5-tethered analogue 29 shown in Figure 4 and observed marked species-dependent differences. Unexpectedly, 29 was found to be much more rapidly cleared when incubated with whole blood of mouse, rat, dog, or monkey, although there was no appreciable difference in stability in human blood between these two compounds. This data was suggestive of the potential for extrahepatic clearance mechanisms for 29 when compared to that for 22 in preclinical species and the reason behind the divergency of predicted and measured clearance in rat. Although the human blood data suggested that this was unlikely to be an issue in patients, a solution was sought in order to achieve suitable exposure in rodents preclinically to assess efficacy and tolerability.
In addition, identification of metabolites formed in vitro for compounds from the tethered series proved informative. Metabolite formation of analogue 28 was assessed following incubation with rodent and human hepatocytes. In human hepatocytes, considerable oxidative metabolism was observed on the tether and quinazoline ring core. In contrast, in rat hepatocytes, the predominant metabolites formed were those of glutathione addition to the acrylamide and assorted products derived from the breakdown of this adduct. This highlighted that the different metabolic fates across species are consistent with the whole blood stability data, where rapid clearance on incubation with rodent blood is indicative of elimination by conjugation to the large amounts of GSH present, but relative stability in human whole blood where this is not a significant elimination route. The 7-membered ring tether 23 showed even greater GSH metabolism in rat hepatocytes consistent with the higher in vitro Clint values, although interestingly, human hepatocytes showed predominantly amide hydrolysis of the acrylamide along with less GSH adduct formation.
In an attempt to address this issue, we turned to our previously successful strategy of methylation of the piperazine adjacent to the acrylamide warhead. In that work, methylation was successful in reducing GSH-mediated clearance by a reduction in Michael acceptor reactivity and steric crowding of the warhead. Unlike in the C3-tethered series that led to 5, methylation on the left as drawn side of the piperazine was poorly tolerated with loss of approximately 26 fold potency, data not shown. However, methylation on the right side was tolerated albeit with a modest decrease in potency, and without any great stereochemical preference for the newly added group shown as 30 and 31. Only analogue 30 had Clint values in an acceptable range, and gratifyingly, the in vivo rat clearance was now reasonable and also in line with predictions at 48 mL/min/kg predicted and observed and with moderate bioavailability. This compound, originally prepared and dosed as a mixture of atropisomers, was separated, and its active atropisomer 32 was dosed in a similar rat PK study exhibiting moderate clearance in agreement with predictions from hepatic scaling and moderate bioavailability. In comparing 29 with its methylated exemplar 32, it is striking how impactful such a simple methylation is in reducing clearance from 962 to 29 mL/min/kg. Despite progress in optimization of rodent PK, this had come at the cost of potency. Indeed, 32 proved to be a relatively modest cellular inhibitor of KRAS function, demonstrating a GI50 of 284 nM in H358 cells which harbor the G12C mutation. Lacking an obvious strategy to further improve potency without also compromising PK, we revisited the 8-membered analogue 28 in an attempt to address the very high in vitro Clint values on this more potent scaffold.
Although 28 had high in vitro human Clint, rodent PK was at least measurable with a low bioavailability of 16 percent associated with a high clearance of 84 mL/min/kg, which was within a twofold range of the predicted clearance of 42 mL/min/kg shown in Table 3. Given that in vitro metabolism studies had highlighted that the different fates of the 7- versus the 8-membered ring lay in metabolism of the tether itself, unsurprising given the additional methylene, we had initially explored ways to limit this through approaches such as deuteration and fluorination of the tether, but none of these strategies proved successful. Interestingly, introduction of the optimized 8-fluoro and 7-phenol groups 33 did yield an appreciable decrease in human microsomal and hepatocyte stability attributable at least in part to a decrease in lipophilicity, although conversely an increase in rat hepatocyte Clint was noted. Despite this, 33 showed reasonable rat PK with a clearance of 25 mL/min/kg with predicted 22 mL/min/kg and moderate bioavailability of 36 percent. Piperazine methylation, adopted previously in 32 to address extrahepatic metabolism, was undertaken to give diastereomers 21 and 34. However, since clearance of 33 appeared to be mainly hepatic metabolic in nature, there was no great expectation that this would yield improvements. Gratifyingly however, R-Me piperazine 21, in which both tether and methyl groups are positioned axially on the top and bottom face of the piperazine, did lead to lowered Clint values despite an increase in LogD, in both human and rat systems. Compound 34, in which the methyl is positioned equatorially, was fortuitously both less potent and more highly cleared than 21. Compound 21, designated Daraxonrasib (RMC-6236), retained potent inhibition of KRASG12C and demonstrated good rat PK with low clearance and high bioavailability. The favorable clearance properties extend to mouse and dog as shown in Table 4. Clearance across two rodent and one non-rodent species is low to moderate at 4 to 31 percent of hepatic blood flow and was generally predicted within twofold from hepatocyte turnover. Direct renal or biliary elimination of unchanged parent compound was not observed in a bile duct cannulated rat elimination study, demonstrating that the observed in vivo clearance for 21 is expected to be hepatic metabolic in nature. Oral bioavailability at low dose in rat and mouse was found to be greater than 70 percent and manifested at 43 percent in dog. Volume of distribution at steady state across species was 0.6 to 1.4 L/kg, which with low to moderate clearance leads to half-lives between 0.6 and 2 hours. It is interesting to note that the preference for, and PK benefits of, having both piperazine groups equatorially mirrors exactly the preference in compound 5, albeit on transposed methylene groups.
Metabolite identification studies in human liver microsomes for 21 identified several core oxidation metabolites, which were not on the tether piperazine ring, highlighting the success of reducing metabolism in this area by addition of the methyl group.
The crystallographic binding mode of 21 (PDB: 7O70) in KRASG12C is shown in Figure 5. As expected from previous SAR and crystal structures, the fluorophenol makes an interaction to Asp69, while the quinazoline N1 atom contacts His95. It is noteworthy that, similar to 28, the unstable water molecule is not displaced and instead the exquisite potency afforded by 21 is a function of excellent protein-ligand shape complementarity and the formation of a favorable network of waters between the O atom of the tether and Arg68. Interestingly, direct stacking to Arg68 is no longer seen for 21. As previously mentioned, the chiral methyl of 21 is oriented axially around the piperazine which results in the restoration of the low-energy chair conformation of the piperazine when viewed against the twist-boat conformation seen in 28. This, alongside the switch to fluorophenol, is likely responsible for the difference in the Arg68 conformation and the resultant binding mode.
One theoretical concern with any agent designed to target non-catalytic cysteine residues in proteins is that of promiscuity. The specificity of 21 by global cysteine reactivity profiling was examined to assess selectivity against the wider cysteine-containing proteome shown in Figure 6. NCI-H358 cells which carry the G12C mutation were treated with 4 micromolar of compound 21 or vehicle for 2 hours. Any remaining unreacted, solvent exposed cysteine residues were then reacted with iodoacetamide desthiobiotin, and proteins were subsequently digested with trypsin. The resulting biotinylated peptides were enriched with streptavidin and analyzed by MS to determine which targets 21 was capable of binding in addition to KRASG12C. In this manner, 21 was found to be highly selective for the engagement of KRASG12C and was also found to reduce the abundance of specific cysteine-containing peptides from both elongation factor 2 (EEF2, cysteine 136) and oxidation resistance protein 1 (OXR1, cysteine 410). The peptide with the largest magnitude change from control was that spanning cysteine 12 of KRASG12C.
In the KRASG12C mutant tumor cell line NCI-H358, 21 was able to potently inhibit the phosphorylation of p90RSK and the growth of this model in a spheroid proliferation assay as shown in Table 5. In the non-KRASG12C tumor cell lines, limited inhibition of either phosphorylation of p90RSK or spheroid proliferation was seen with 21 however, and potency in these cells was reduced at least 100-fold compared to that observed in NCI-H358 cells. Daily oral treatment of mice with 21 at 100, 20, and 4 mg/kg was well tolerated and inhibited the growth of KRASG12C mutant MIA PaCa-2 xenograft tumors, with significant tumor regressions at the higher doses shown in Figure 7. Tumors from mice treated with 21 showed a robust, dose-dependent decrease in the expression of DUSP6, a well-characterized transcript regulated by RAS/MAPK pathway activity at 3 and 6 hours post-dose. The data in Figure 7 demonstrate a clear PD-efficacy relationship for 21 in the MIA PaCa-2 model, with increased pathway inhibition corresponding to increased efficacy. Taken together, these data indicate that 21 is a potent and selective inhibitor of KRASG12C.
Conclusions
The discovery of compound 21, a potent and selective KRASG12C inhibitor for clinical development designated Daraxonrasib (RMC-6236), has been described. An initial key step in its discovery was the application of a conformational tether to lock the piperazine group at C4 back to C5 of a quinazoline ring to derive a tetracyclic quinoline which resulted in significant increases in potency dependent on both the ring size and stereochemistry that could be rationalized from structural and conformational analyses. Overcoming an observed extrahepatic clearance mechanism in rodents linked to the addition of glutathione to the warhead was achieved by addition of a single methyl group adjacent to the warhead. The potency gains obtained from this strategy may be explained by exquisite conformational locking of the bioactive pose which minimizes the entropy loss upon binding and maximizes protein-ligand complementarity. Across species, the compound demonstrated low clearance and high bioavailability. Compound 21 was highly selective for reaction with the cysteine in KRASG12C over other cysteine-containing proteins and had a selective antiproliferative phenotype in cancer cells bearing the G12C mutation that led to strong tumor regressions in a mouse xenograft disease model.